¿Qué es La Atrofia Muscular Espinal?
La Atrofia Muscular Espinal (AME) es una enfermedad de causa genética que predominantemente afecta a pacientes en edad pediátrica y que tiene como manifestación principal la debilidad muscular progresiva y finalmente la atrofia de estos músculos.

Sus síntomas
Como se mencionó, la debilidad muscular y la atrofia son característicos en esta enfermedad. Según la edad de comienzo de los síntomas y los logros motores que puedan alcanzar los pacientes (por ejemplo, sostener la cabeza, sentarse, caminar) se ha clasificado en distintos tipos:





AME tipo I. Es la forma más frecuente de la enfermedad. Pueden comenzar a tener síntomas luego de las primeras semanas del nacimiento. Habitualmente es percibida por los padres la dificultad en la succión, para tragar o la dificultad para respirar. Estos pacientes no logran sostener la cabeza en la mayoría de los casos y no van a lograr sentarse. Característicamente, a medida que evoluciona la enfermedad, van a necesitar apoyo para respirar, como es la conexión a una ventilación asistida y apoyo para alimentarse, ya sea con una sonda naso gástrica o la alimentación directa por gastrostomía. (Inserción de un catéter directamente en el estómago para la alimentación). El diagnóstico lo más rápido y posible es fundamental para poder brindar un tratamiento.
AME tipo II. Sigue en frecuencia a la anterior, suelen comenzar con síntomas luego de cumplido el sexto o séptimo mes de vida. Presentan debilidad generalizada pero predominantemente en los miembros inferiores y no logran caminar en forma independiente.
AME tipo III y tipo IV. Son mucho menos frecuentes que las anteriores. En ambos casos podrían lograr deambular, pero esta habilidad puede perderse a medida que progresa la enfermedad.
La causa de la debilidad muscular y la atrofia
Los músculos se mueven gracias a un impulso eléctrico proveniente desde la corteza cerebral, que viaja a través de la médula espinal y que finalmente “ordena” al músculo para que se contraiga. En este proceso, la neurona que emite la señal desde el cerebro se comunica con una segunda neurona ubicada en la médula, denominada motoneurona inferior, y finalmente esta “ordena” la contracción muscular.
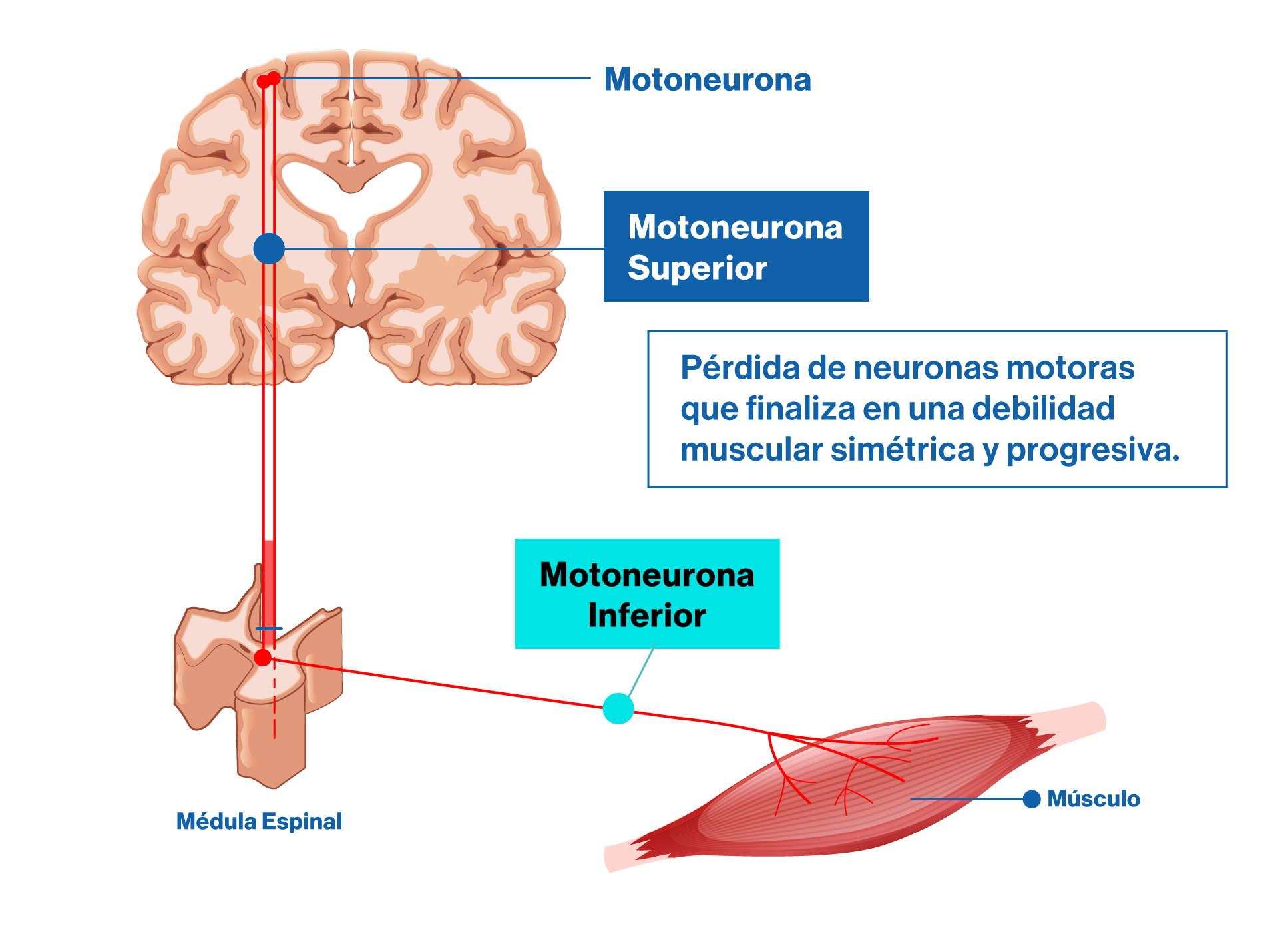
En AME, la motoneurona inferior “muere” y por lo tanto no ejecuta la orden de contracción del músculo al que inerva. Luego, el músculo, por falta de estímulo termina atrofiándose y perdiendo función.
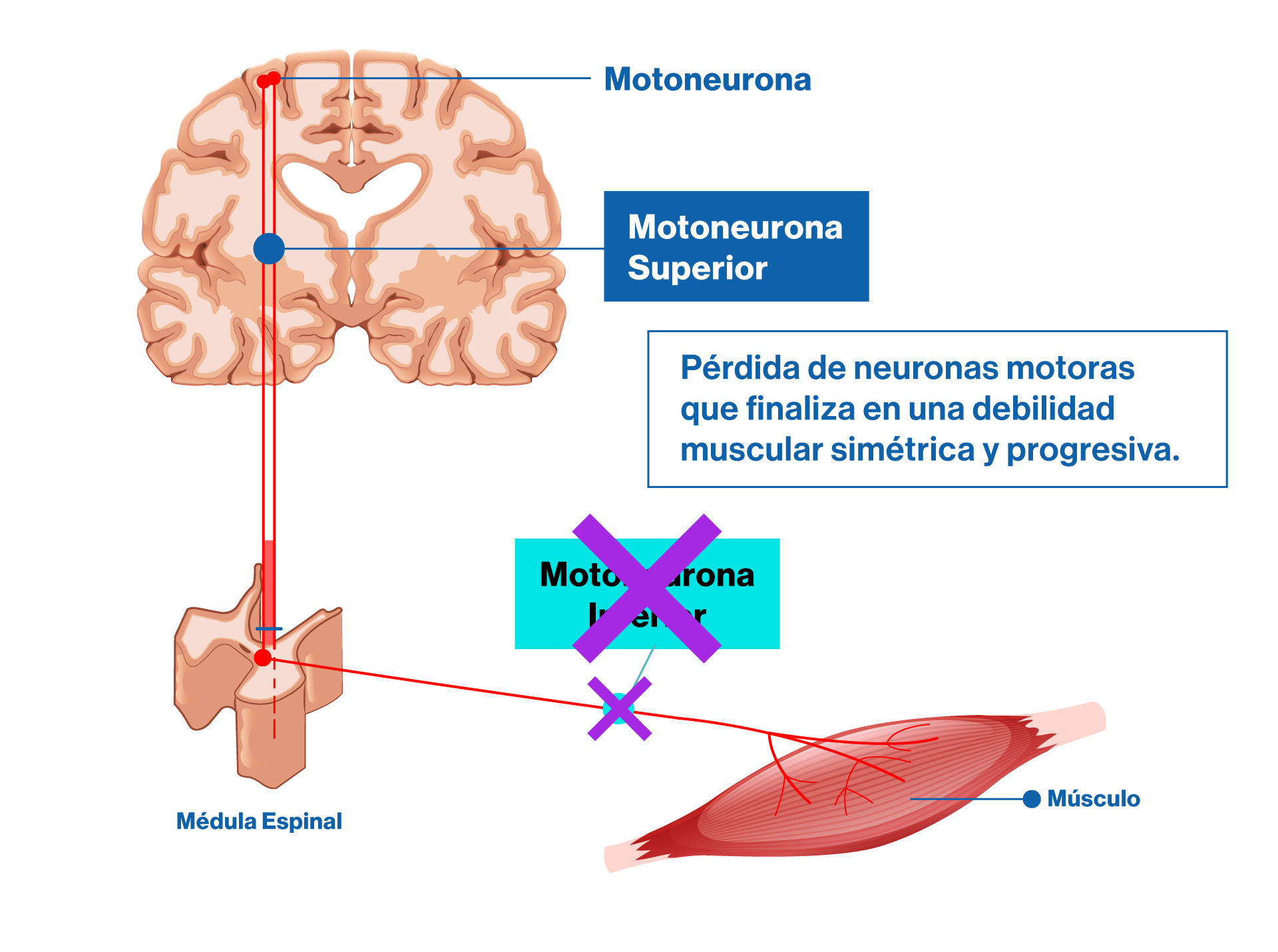
La causa de la pérdida de motoneuronas radica en la deficiencia que tienen estos pacientes de una proteína denominada “Survival Motor Neuron” o proteína SMN. Si bien no es del todo bien comprendida cuál es la función de esta proteína, está claro que su ausencia lleva a la pérdida de estas neuronas, de allí el nombre de “Survival Motor Neuron” o “proteína de supervivencia neuronal” en su traducción al español.
Genética de la enfermedad y herencia
Como se mencionó la causa fundamental de la AME, radica en la deficiencia de la proteína SMN. Esta deficiencia tiene una causa genética. Esta proteína se produce gracias a la existencia de un gen denominado gen SMN1 y a otro gen secundario o de respaldo, denominado gen SMN2. El gen SMN1 es el responsable de la mayor producción de proteína SMN, mientras que el gen SMN2 produce solo el 10% de esta proteína.
La mayoría de los humanos contamos con 2 copias del gen SMN1 y otras 2 copias del gen SMN2. Algunas personas tienen variantes en la cantidad de copias del gen SMN2, pudiendo tener 1,3, 4 o más copias, habiéndose descripto casos con hasta 6 copias. La cantidad de copias del gen SMN2 no tiene ninguna implicancia para las personas sin AME, pero es relevante (como se explicará luego) para los pacientes con la enfermedad.
Característicamente, los pacientes con AME poseen mutaciones en el gen SMN1, y por lo tanto, la producción de proteína SMN es insuficiente para mantener “vivas” a las motoneuronas. Como se ha mencionado, una pequeña cantidad de proteína puede ser producida por el gen de respaldo, el gen SMN2. Esta cantidad, va a depender del número de copias que presente el paciente, y el pronóstico será más favorable cuanta más cantidad de copias posea. Así, por ejemplo, la mayoría de los pacientes con AME tipo I poseen una o dos copias, los AME tipo II poseen dos o tres copias, los AME tipo III poseen tres o cuatro copias y los pacientes con AME tipo IV suelen tener tres, cuatro o más copias del gen SMN2.
Herencia
Los pacientes con AME, reciben una copia afectada del gen SMN1 de cada uno de sus padres. Los padres son denominados “portadores” ya que tienen una de las dos copias del gen SMN1 alterada y la otra “sana” y por lo tanto no manifiestan la enfermedad. Según estudios, se conoce que aproximadamente 1 de cada 50 personas es portador.

Diagnóstico
El diagnóstico de certeza de la enfermedad se realiza a través del estudio genético del paciente, buscando la mutación en el gen SMN1. Adicionalmente se estudia el número de copias del gen SMN2, en este caso no para diagnosticar, sino solo para tener una referencia aproximada del comportamiento que pueda tener la misma.
“Material informativo entregado por el Programa de Soporte a Pacientes, elaborado para fomentar condiciones de vida saludables”.

Tratamiento para AME

¿Cómo preparar a nuestros hijos
ante una extracción de sangre?
